
Introduction
In today’s post, I will present the synthesis of Benzoin through a thiamine-catalyzed condensation reaction between two benzaldehyde molecules. The overall reaction can be represented as follows:

Where thiamine, also known as vitamin B1, is used as a catalyst. The structure of the thiamine molecule, in its hydrochloride salt form, is the following:

Under alkaline conditions, thiamine can be liberated in its free-base form and, with an excess of base, the molecule can be deprotonated by removing the hydrogen between the sulfur and nitrogen atoms of the thiazole ring. This leads to the formation of a carbene-like structure according to the reaction:

The formed ylide structure can attack a benzaldehyde molecule giving rise to an activated intermediate in which the proton, formally belonging to the aldehyde function, assumes an acidic character. Such proton can then be abstracted by a base generating an enamine-like moiety involving the thiazole nitrogen. The generated structure has a nucleophilic character and can attack another benzaldehyde molecule forming a new carbon-carbon bond. After the reaction, deprotonation of the hydroxide group at the carbon linked to the thiazole function liberates the thiamine carbene catalyst and regenerates the original carbonyl function. The product of the reaction is the desired benzoin product. The overall reaction mechanism can be represented as follows:

The original procedure followed for the reaction presented in this post has been taken from Joaquín Isac-García, José A. Dobado, Francisco G. Calvo-Flores, Henar Martínez-García, Experimental Organic Chemistry: Laboratory Manual, Chapter 9: Advanced Organic Synthesis Experiments, Academic Press, 2016, Pages 291-352, ISBN 9780128038932, (https://doi.org/10.1016/B978-0-12-803893-2.50009-7)
Experimental part
In a 100ml flat bottom flask equipped with a magnetic stir bar, 0.7g of thiamine hydrochloride (2.08mmol, 0.053eq.) were dissolved in 1.5mL of deionized water. 6mL of 95% ethanol were added to the solution and the resulting mixture was chilled in an ice bath until its temperature dropped below 5°C. On the side, a 3M sodium hydroxide solution was prepared by dissolving 1.8g (45mmol) of the base in 15mL of deionized water. The sodium hydroxide solution was chilled in an ice bath and, when cold, 1.5mL (4.5mmol, 0.11eq.) of it were added, under constant stirring, to the reaction mixture. The addition was made in small portions over the course of 10 minutes never exceeding the 6°C mark. The reaction mixture assumed, at first, a dark yellow color that, upon standing, lightened slightly. 4mL of benzaldehyde (4.2g, 39mmol, 1.0eq.) were added to the reaction mixture, the pH was checked with a litmus paper and found to be around 7. The pH was adjusted to above 8 by adding a few additional drops of sodium hydroxide solution. The solution was removed from the ice bath and heated at 65°C for 1h in a water bath. During the reaction, a distinct sulfury smell was noted and the color of the mixture darkened. At the end of the heating, the ambered yellow solution was left to cool down and a precipitate was observed. The reaction mixture was cooled in an ice bath and the solid, recovered using vacuum filtration, was rinsed with cool deionized water. A total of 2.15g of not perfectly dried solid were produced corresponding to a yield lower than 51%. The recovered solid had a light yellow/cream color and, judging just by its smell, contained trace amounts of unreacted benzaldehyde. The solid was recrystallized from a boiling ethanol-water mixture, recovered by vacuum filtration, rinsed with deionized water, and dried over anhydrous calcium chloride. A total of 1.51g of white translucent needle-like crystals were recovered corresponding to a final yield of 36%.
The terrible yield can be ascribed to the reaction not going to completion probably due to insufficiently long reaction times or sub-optimal reaction conditions. As a matter of fact, after the water rinsings, the crude filtrate presented a significant quantity of unreacted benzaldehyde that phased out as an oily liquid. Yield can probably be improved with longer reaction times and by better tuning pH and temperature. If available, following the reaction with TLC would be advisable. In the future, I plan to repeat the experiment to improve the yield and I will post an update if better reaction conditions are found.

Product characterization
The 1H, 13C, and HSQC spectra have been recorded using a 500MHz instrument dissolving the sample in chloroform-d.
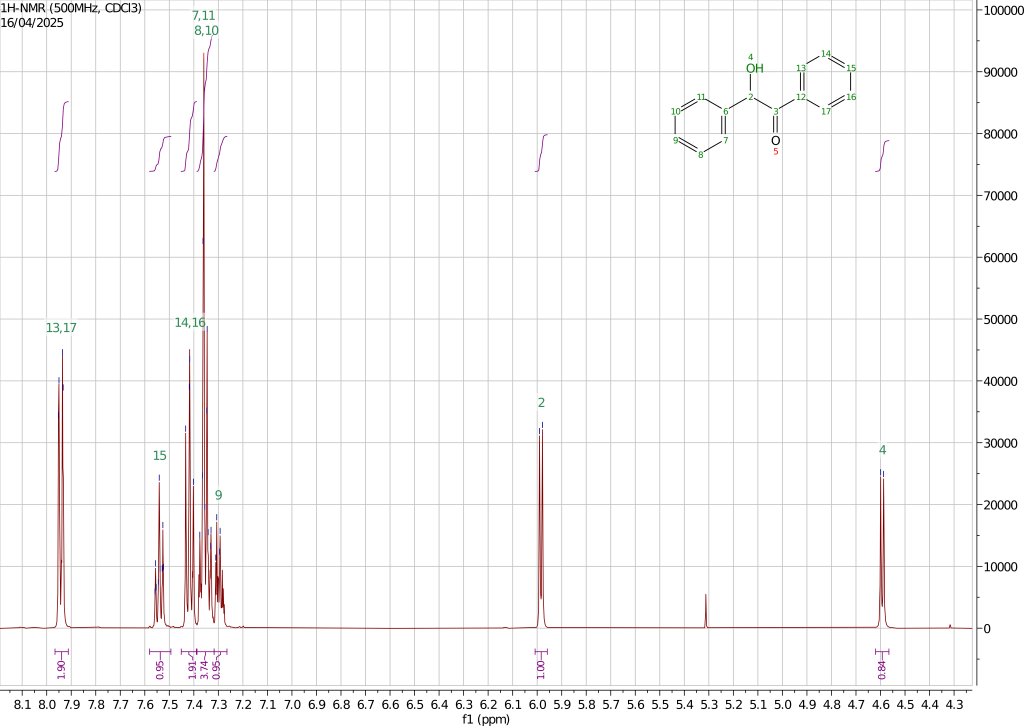
The proton spectrum is consistent with the expected compound. Two doublets coupled by a constant of 6.1Hz are observed at 4.59 and 5.98ppm and are associated with the hydroxy proton and the adjacent CH group on the alkyl chain. The aromatic protons are responsible for the signals observed in the region between 8 and 7.2ppm. Thanks to the electron-withdrawing properties of the carbonyl group, the protons of the phenyl ring directly connected to it are characterized by larger chemical shifts. As expected from chemical intuition, the protons in ortho to the carbonyl functions are characterized by the highest chemical shift of 7.94ppm followed by the proton in para at 7.54ppm and those in meta at 7.42. The splitting pattern is consistent with this assignment with the protons in ortho showing as a doublet of doublets showing a 8.3-8.4Hz coupling with the ones in meta and a smaller 1.2-1.3Hz coupling with the more distant proton in para. The para proton shows a triplet-like structure with a coupling of 7.4-7.5Hz with the meta ones and a secondary splitting due to the coupling to the ortho protons. The meta protons show a triplet-like structure consistent with the previously discussed coupling constants. The protons of the second phenyl ring appear as a more complex multiplet pattern where four protons give rise to a multiplet between 7.39-7.32ppm and one, possibly para one, at 7.32-7.26ppm.
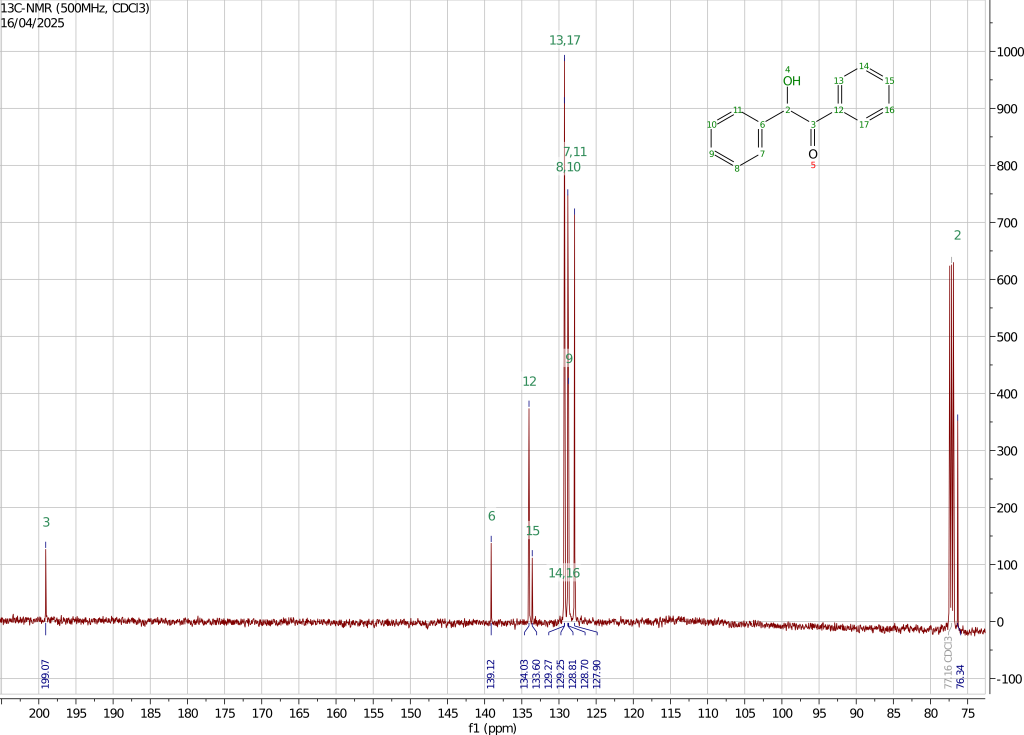
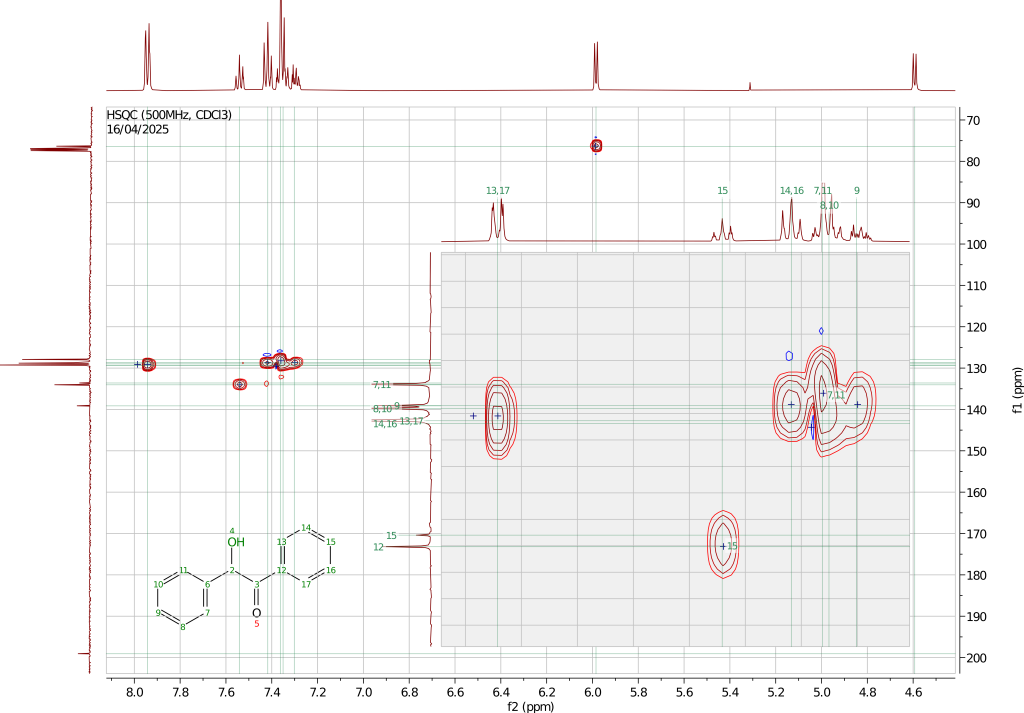
The 13C spectrum is also consistent with the presented structure with the carbonyl carbon shifted at 199.07ppm and the CH carbon at 76.34ppm. The carbons of the aromatic rings give rise to the remaining eight signals two of which are almost coincident at 129.27 and 125.25ppm. Assigning these peaks is not trivial. By looking at the HSQC spectrum it is easy to spot the two carbon signals associated with the quaternary aromatic carbons that, not being directly coupled with a proton, do not show in the HSQC spectrum.
Very nice blog, congrats!
This reaction gives around 70% yield if you don’t heat it and just let it sit for a few days at room temperature.
Hi Martin, thank you so much for your kind words and for the excellent suggestion! I just posted a follow-up in which I tested exactly that; the yield improved significantly by raising the pH and running the reaction at ambient temperature. I haven’t managed to obtain a 70% yield yet, but I probably lost part of the product during recrystallization.